CNR Bioinformatics Workshop
4. Visualization
4.1 Load libraries
library(ggplot2)
library(Seurat)
library(dplyr)
4.2 Load data
DEX <- read.csv("DEX.csv")
head(DEX)
| X | p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | cluster | gene | |
|---|---|---|---|---|---|---|---|---|
| <chr> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <chr> | |
| 1 | THSD7A | 6.045978e-196 | 1.869736 | 0.936 | 0.344 | 9.984329e-192 | 0 | THSD7A |
| 2 | CDH7 | 8.096795e-188 | 1.887889 | 0.677 | 0.088 | 1.337105e-183 | 0 | CDH7 |
| 3 | CADM1 | 9.910354e-165 | 1.451553 | 0.961 | 0.594 | 1.636596e-160 | 0 | CADM1 |
| 4 | NTS | 1.028108e-156 | 2.381170 | 0.753 | 0.192 | 1.697817e-152 | 0 | NTS |
| 5 | LINC00643 | 6.132768e-156 | 1.219894 | 0.821 | 0.219 | 1.012765e-151 | 0 | LINC00643 |
| 6 | CNTNAP2 | 1.002729e-146 | 1.338118 | 0.975 | 0.578 | 1.655906e-142 | 0 | CNTNAP2 |
df0 <- readRDS("df0.rds")
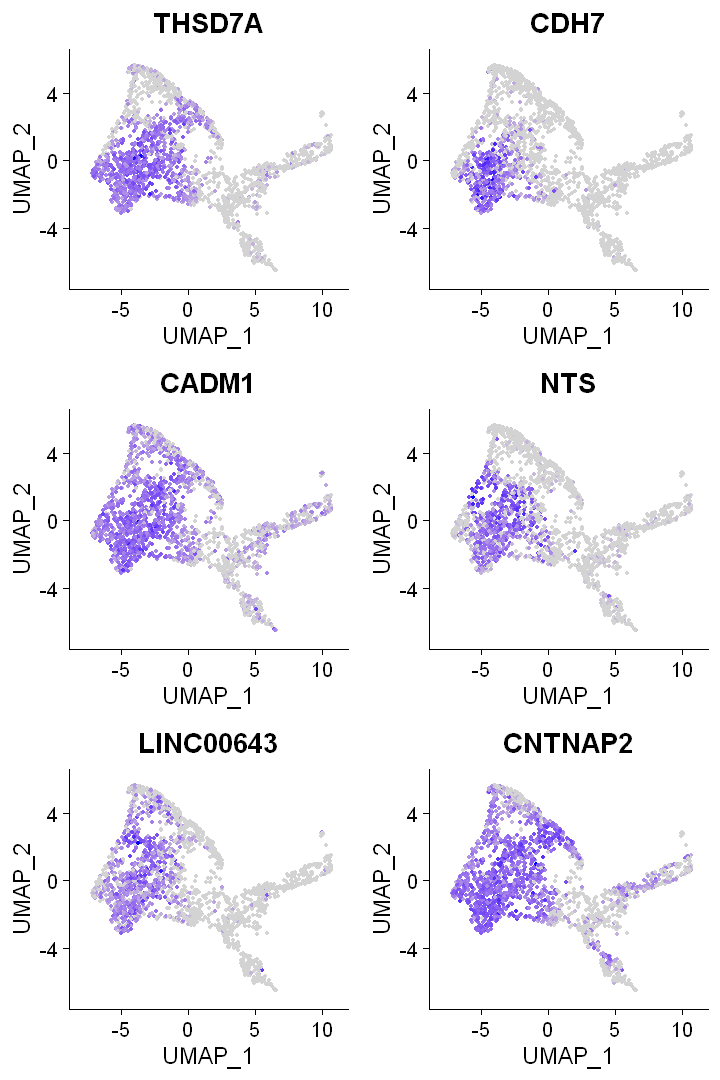
4.3 Feature plot
genes <- head(DEX)$gene
Make Feature plots with Seurat function FeaturePlot
options(repr.plot.width = 6, repr.plot.height = 9)
FeaturePlot(df0, features = genes)

Use NoLegend function to remove function
options(repr.plot.width = 6, repr.plot.height = 9)
FeaturePlot(df0, features = genes) + NoLegend()

Use combine = F to create a list of plots that you can modify individually
plist <- FeaturePlot(df0, features = genes, combine = F)
typeof(plist)
‘list’
Use apply to change each object from the list one at a time
plist <- lapply(plist, function(x){
x = x + NoLegend()
})
Use do.call and gridExtra to present figures
options(repr.plot.width = 6, repr.plot.height = 9)
do.call(gridExtra::grid.arrange, c(plist, ncol = 2))
Remove axes and change color scale
plist <- lapply(plist, function(x){
x = x + scale_color_gradientn(colours = c("grey", RColorBrewer::brewer.pal(9, "YlOrRd"))) +
theme_void()
return(x)
})
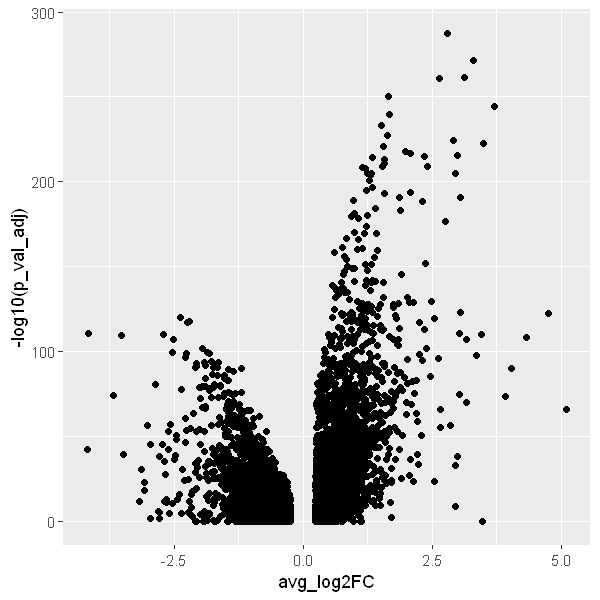
4.4 Volcano plot
range(DEX$avg_log2FC)
<ol class=list-inline><li>0.250067637009388</li><li>5.11402342406602</li></ol>
options(repr.plot.width = 5, repr.plot.height = 5)
ggplot(DEX, aes(x = avg_log2FC, y = -log10(p_val_adj))) +
geom_point()
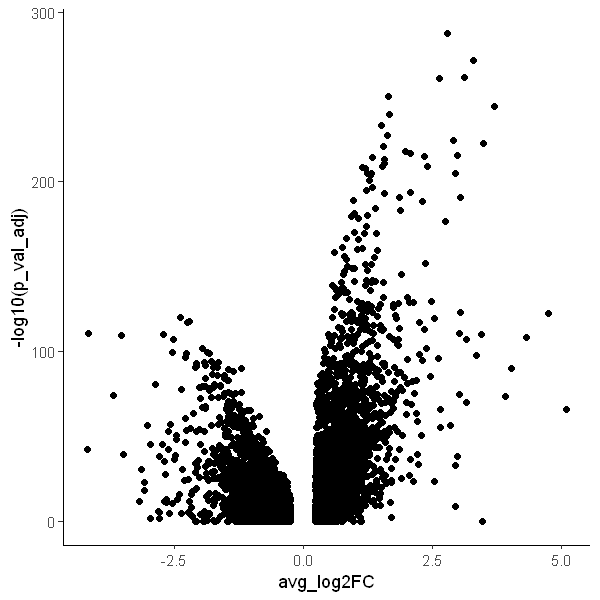
Use different ggplot2 themes
options(repr.plot.width = 5, repr.plot.height = 5)
ggplot(DEX, aes(x = avg_log2FC, y = -log10(p_val_adj))) +
geom_point() +
theme_classic()
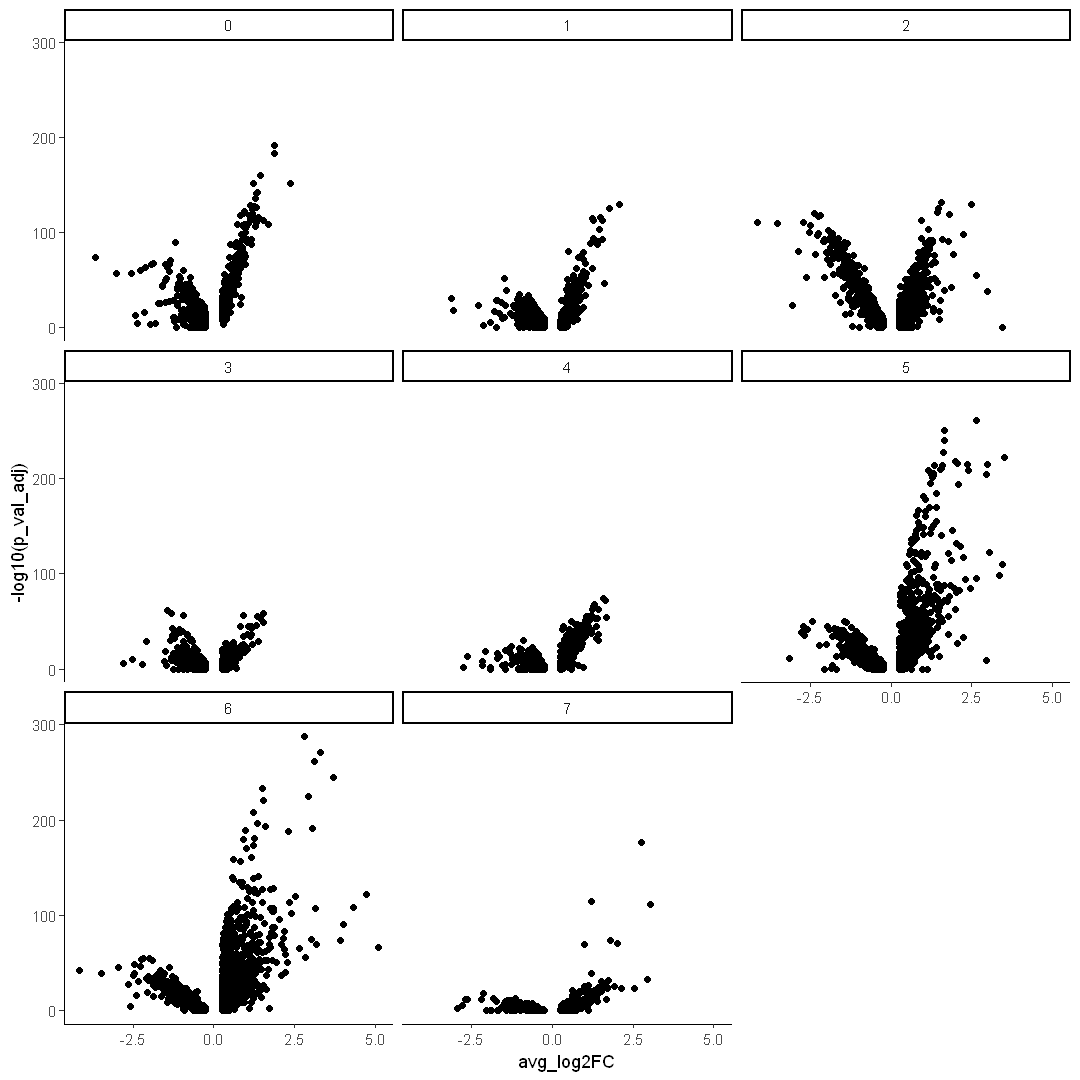
Use facet wrap to separate different clusters into individual plots
options(repr.plot.width = 9, repr.plot.height = 9)
ggplot(DEX, aes(x = avg_log2FC, y = -log10(p_val_adj))) +
geom_point() +
facet_wrap(~cluster) +
theme_classic()
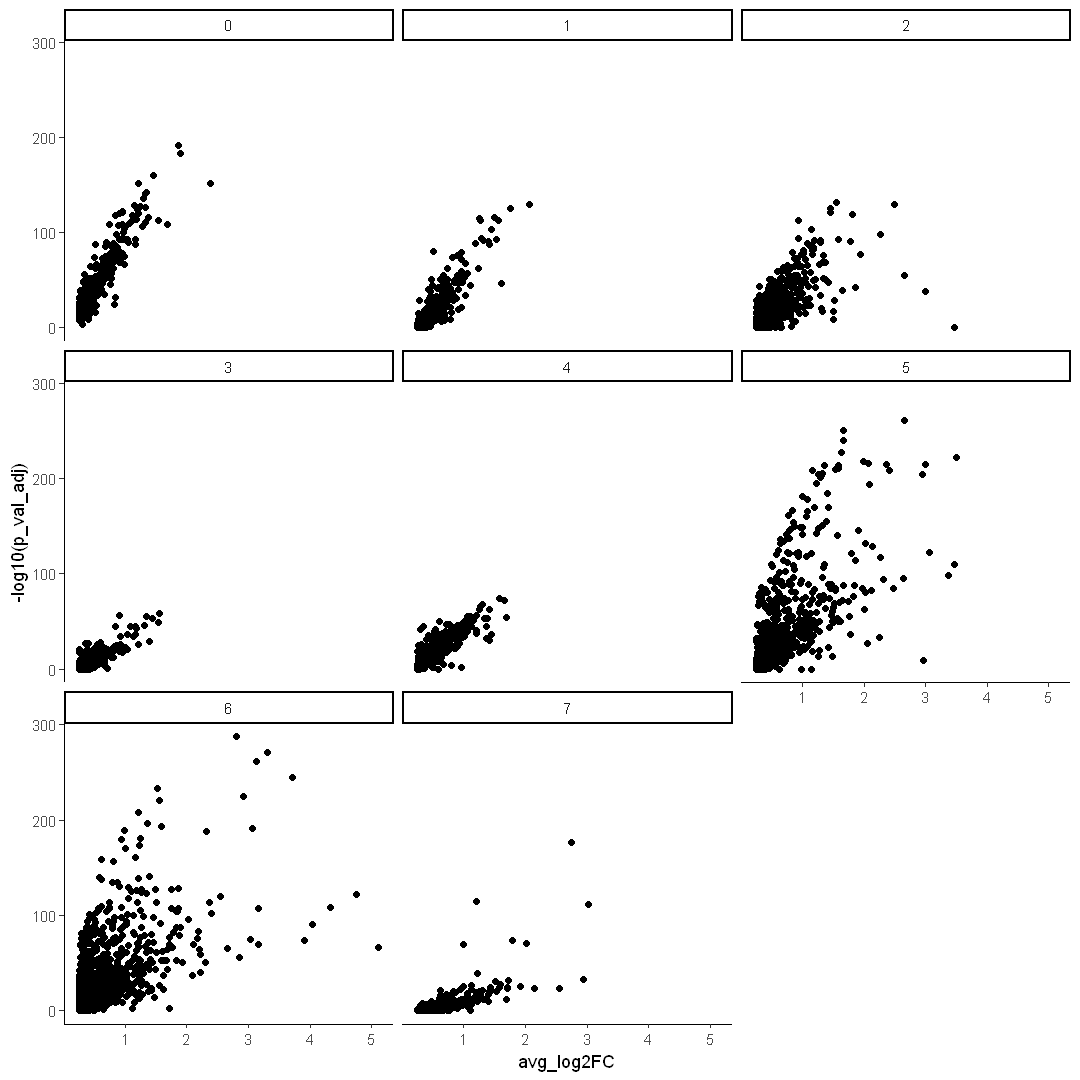
Use dplyr to subset data
options(repr.plot.width = 9, repr.plot.height = 9)
ggplot() +
DEX %>%
filter(avg_log2FC > 0) %>%
geom_point(mapping = aes(x = avg_log2FC, y = -log10(p_val_adj)), data = .) +
facet_wrap(~cluster) +
theme_classic()
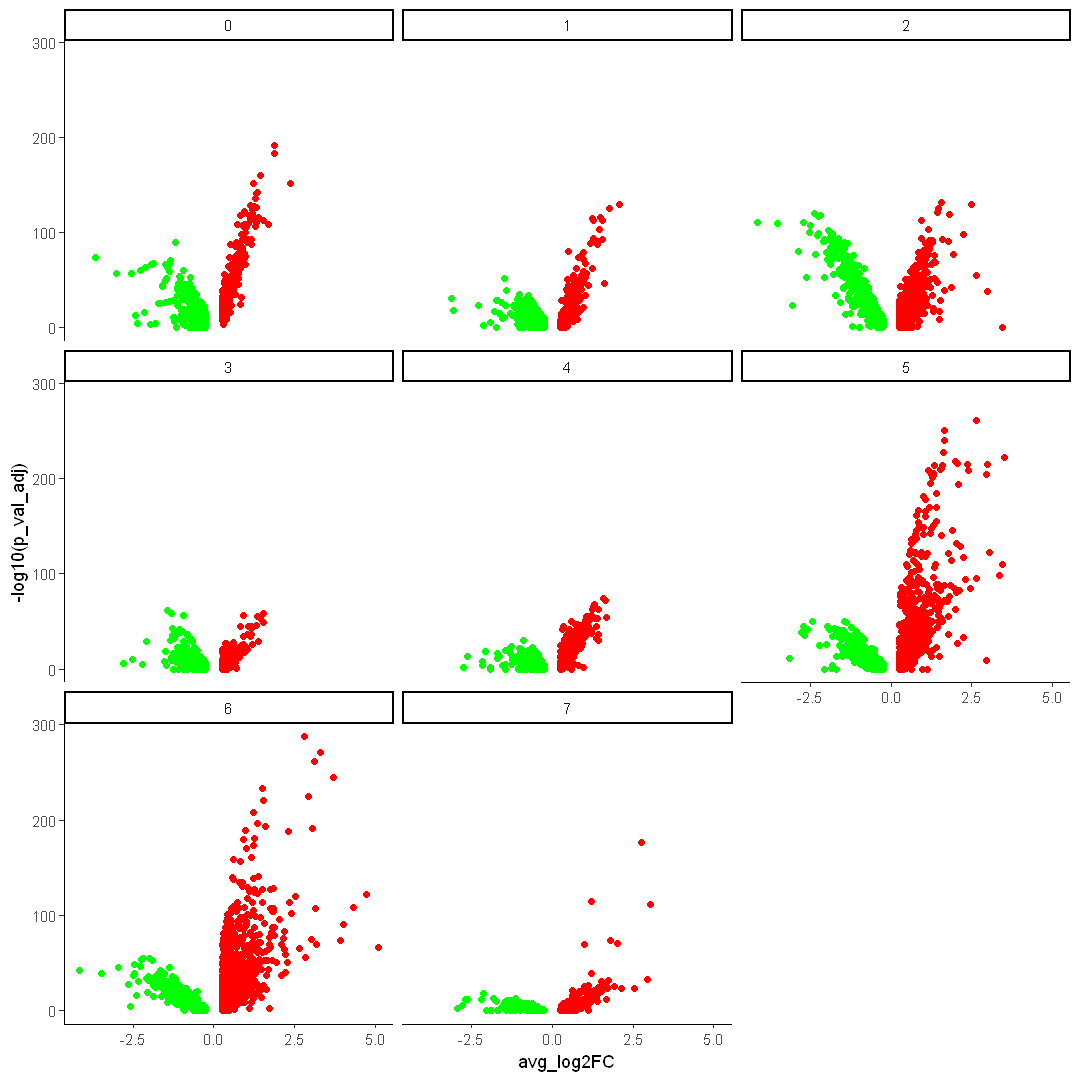
Use different colors
options(repr.plot.width = 9, repr.plot.height = 9)
ggplot() +
DEX %>%
filter(avg_log2FC > 0) %>%
geom_point(mapping = aes(x = avg_log2FC, y = -log10(p_val_adj)), data = ., color = "red") +
DEX %>%
filter(avg_log2FC < 0) %>%
geom_point(mapping = aes(x = avg_log2FC, y = -log10(p_val_adj)), data = ., color = "green") +
facet_wrap(~cluster) +
theme_classic()
4.5 Violin plots
genes
<ol class=list-inline><li>‘THSD7A’</li><li>‘CDH7’</li><li>‘CADM1’</li><li>‘NTS’</li><li>‘LINC00643’</li><li>‘CNTNAP2’</li></ol>
data.to.plot <- as.data.frame(t(as.matrix(df0@assays$RNA@data[genes,])))
data.to.plot$cluster <- df0$seurat_clusters
head(data.to.plot)
| THSD7A | CDH7 | CADM1 | NTS | LINC00643 | CNTNAP2 | cluster | |
|---|---|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> | |
| AAACCCAGTCTCAGGC-1 | 2.5219109 | 1.125659 | 1.416672 | 3.4730135 | 0.7135008 | 2.521911 | 0 |
| AAACCCAGTGACTATC-1 | 1.7966931 | 0.000000 | 2.490332 | 0.6961103 | 1.3907357 | 1.390736 | 4 |
| AAACCCATCCCATTTA-1 | 0.0000000 | 0.000000 | 0.000000 | 1.2377887 | 0.0000000 | 1.774268 | 4 |
| AAACCCATCTGAGAAA-1 | 0.6287137 | 0.000000 | 1.011744 | 0.0000000 | 0.0000000 | 1.011744 | 4 |
| AAACGAACAATCTGCA-1 | 0.0000000 | 0.000000 | 0.000000 | 0.0000000 | 0.0000000 | 0.000000 | 3 |
| AAACGCTCAAATGGAT-1 | 1.1773758 | 0.000000 | 1.384908 | 4.1090559 | 1.8310291 | 1.703236 | 4 |
data.to.plot2 <- reshape2::melt(data.to.plot, id.vars = c("cluster"))
head(data.to.plot2)
| cluster | variable | value | |
|---|---|---|---|
| <fct> | <fct> | <dbl> | |
| 1 | 0 | THSD7A | 2.5219109 |
| 2 | 4 | THSD7A | 1.7966931 |
| 3 | 4 | THSD7A | 0.0000000 |
| 4 | 4 | THSD7A | 0.6287137 |
| 5 | 3 | THSD7A | 0.0000000 |
| 6 | 4 | THSD7A | 1.1773758 |
options(repr.plot.width = 9, repr.plot.height = 3)
ggplot(data.to.plot2, aes(x = variable, y = value)) +
geom_violin()
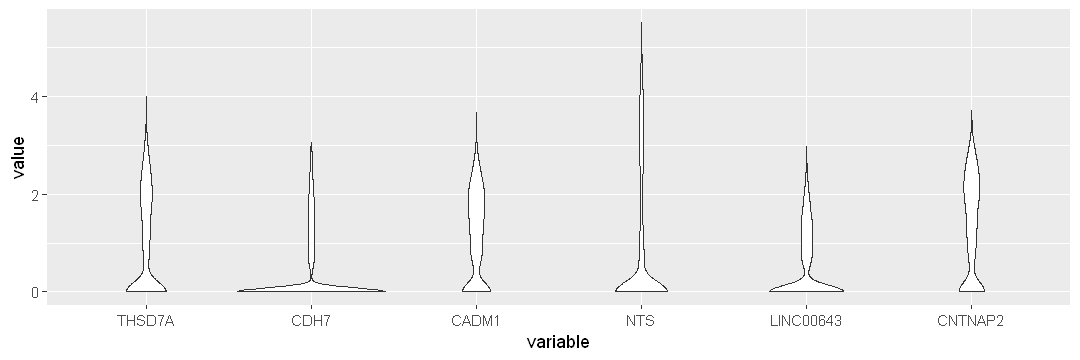
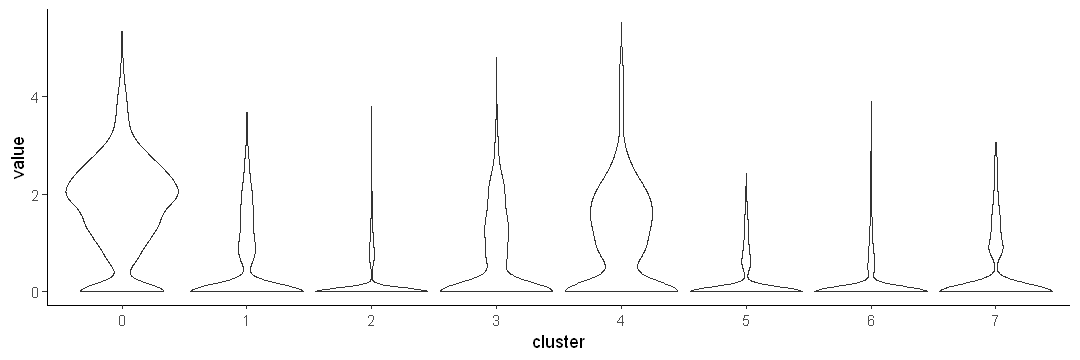
Use scale = width
options(repr.plot.width = 9, repr.plot.height = 3)
ggplot(data.to.plot2, aes(x = cluster, y = value)) +
geom_violin(scale = "width") +
theme_classic()
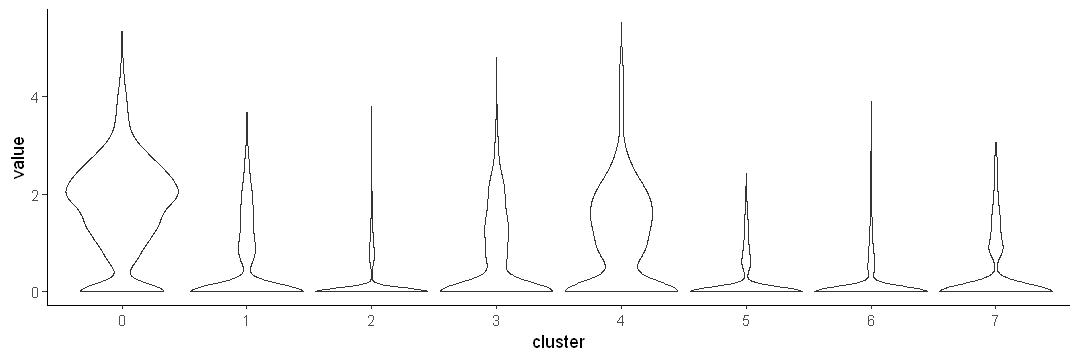
Use scale = width
options(repr.plot.width = 9, repr.plot.height = 3)
ggplot(data.to.plot2, aes(x = cluster, y = value)) +
geom_violin(scale = "width") +
theme_classic()
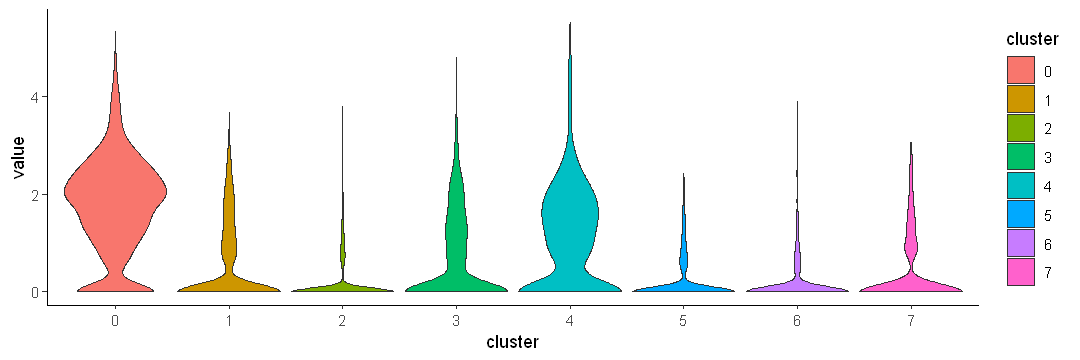
Use fill to add color
options(repr.plot.width = 9, repr.plot.height = 3)
ggplot(data.to.plot2, aes(x = cluster, y = value, fill = cluster)) +
geom_violin(scale = "width") +
theme_classic()
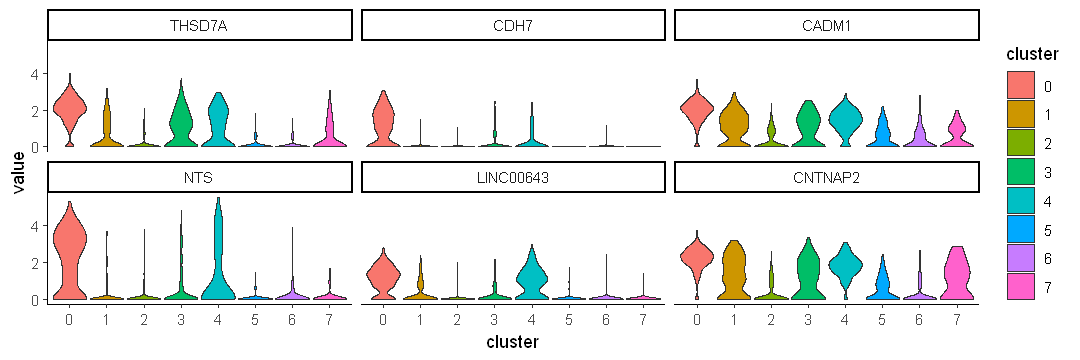
Use facet wrap to show each gene individually
options(repr.plot.width = 9, repr.plot.height = 3)
ggplot(data.to.plot2, aes(x = cluster, y = value, fill = cluster)) +
geom_violin(scale = "width") +
facet_wrap(~variable) +
theme_classic()
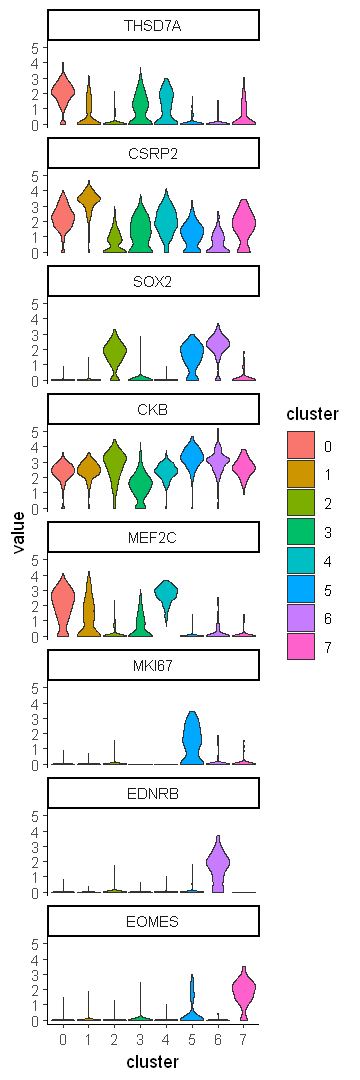
Pick the most significant DEX gene from each cluster
DEX %>%
group_by(cluster) %>%
top_n(-p_val_adj, n = 1)
| X | p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | cluster | gene |
|---|---|---|---|---|---|---|---|
| <chr> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <chr> |
| THSD7A | 6.045978e-196 | 1.869736 | 0.936 | 0.344 | 9.984329e-192 | 0 | THSD7A |
| CSRP2 | 4.887031e-134 | 2.070127 | 0.994 | 0.814 | 8.070443e-130 | 1 | CSRP2 |
| SOX2.2 | 1.213735e-136 | 1.551859 | 0.892 | 0.186 | 2.004362e-132 | 2 | SOX2 |
| CKB.2 | 9.850717e-67 | -1.437922 | 0.861 | 0.982 | 1.626747e-62 | 3 | CKB |
| MEF2C.3 | 6.308674e-79 | 1.580887 | 1.000 | 0.508 | 1.041814e-74 | 4 | MEF2C |
| MKI67 | 1.433349e-265 | 2.652864 | 0.690 | 0.011 | 2.367033e-261 | 5 | MKI67 |
| EDNRB.1 | 2.275104e-292 | 2.798285 | 0.839 | 0.019 | 3.757107e-288 | 6 | EDNRB |
| EOMES.1 | 1.183226e-181 | 2.753743 | 0.899 | 0.037 | 1.953980e-177 | 7 | EOMES |
DEX %>%
group_by(cluster) %>%
top_n(-p_val_adj, n = 1) %>%
select(gene)
[1m[22mAdding missing grouping variables: `cluster`
| cluster | gene |
|---|---|
| <int> | <chr> |
| 0 | THSD7A |
| 1 | CSRP2 |
| 2 | SOX2 |
| 3 | CKB |
| 4 | MEF2C |
| 5 | MKI67 |
| 6 | EDNRB |
| 7 | EOMES |
gene <- DEX %>%
group_by(cluster) %>%
top_n(-p_val_adj, n = 1) %>%
select(gene)
[1m[22mAdding missing grouping variables: `cluster`
data.to.plot <- as.data.frame(t(as.matrix(df0@assays$RNA@data[gene$gene,])))
data.to.plot$cluster <- df0$seurat_clusters
data.to.plot2 <- reshape2::melt(data.to.plot, id.vars = c("cluster"))
options(repr.plot.width = 3, repr.plot.height = 9)
ggplot(data.to.plot2, aes(x = cluster, y = value, fill = cluster)) +
geom_violin(scale = "width") +
facet_wrap(~variable, ncol = 1) +
theme_classic()
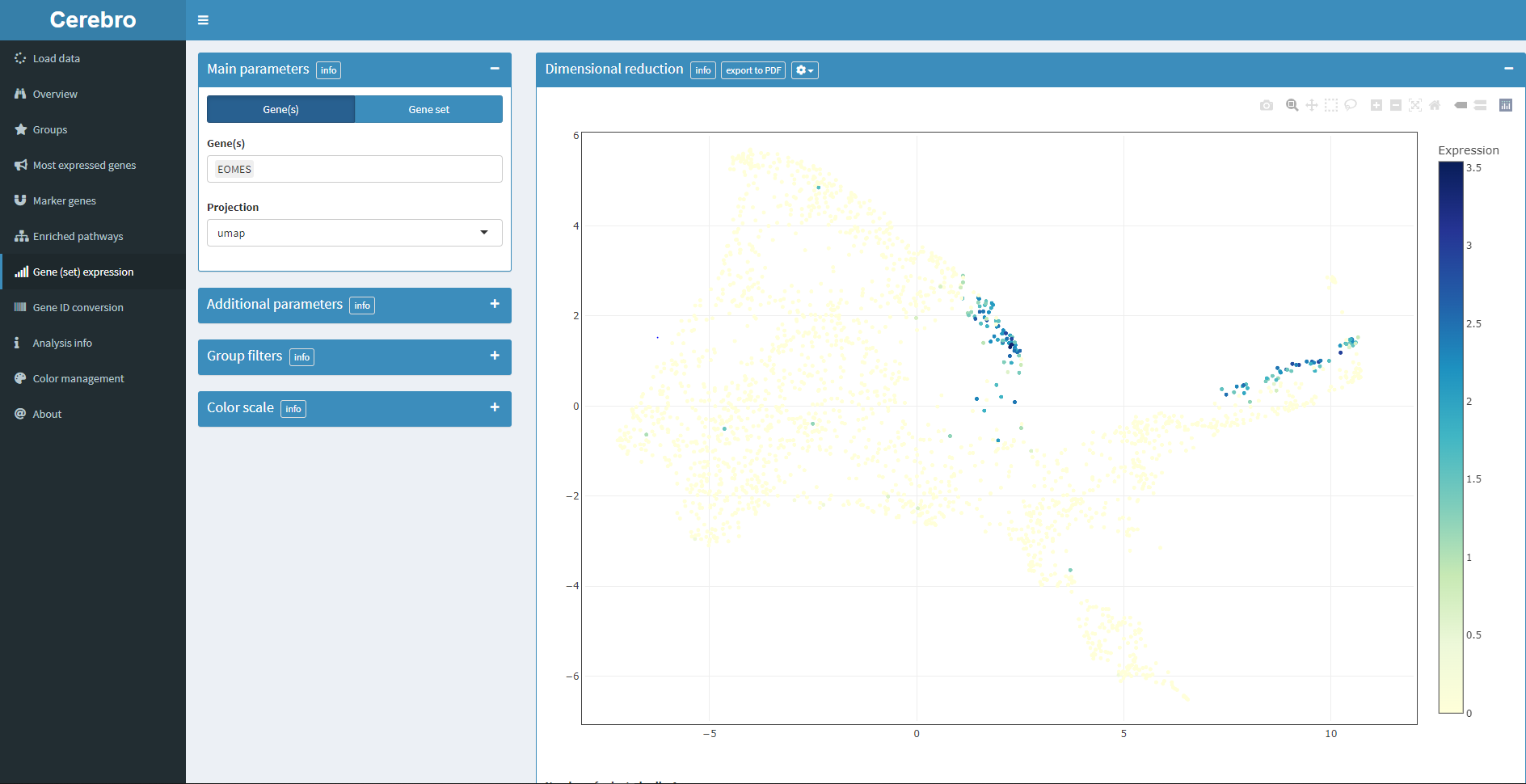
4.6 Install and run Cerebro
install.packages("remotes")
package 'remotes' successfully unpacked and MD5 sums checked
The downloaded binary packages are in
C:\Users\ZLI2\AppData\Local\Temp\3\Rtmp6hrUI3\downloaded_packages
BiocManager::install("romanhaa/cerebroApp")
'getOption("repos")' replaces Bioconductor standard repositories, see
'?repositories' for details
replacement repositories:
CRAN: https://cran.r-project.org
Bioconductor version 3.14 (BiocManager 1.30.18), R 4.1.3 (2022-03-10)
Installing github package(s) 'romanhaa/cerebroApp'
Downloading GitHub repo romanhaa/cerebroApp@HEAD
Rcpp (1.0.8.3 -> 1.0.9) [CRAN]
later (1.2.0 -> 1.3.0) [CRAN]
Installing 2 packages: Rcpp, later
There is a binary version available but the source version is later:
binary source needs_compilation
Rcpp 1.0.8.3 1.0.9 TRUE
Binaries will be installed
package 'Rcpp' successfully unpacked and MD5 sums checked
Warning message:
"cannot remove prior installation of package 'Rcpp'"
Warning message in file.copy(savedcopy, lib, recursive = TRUE):
"problem copying C:\Users\ZLI2\Miniconda3\envs\myEnv\lib\R\library\00LOCK\Rcpp\libs\x64\Rcpp.dll to C:\Users\ZLI2\Miniconda3\envs\myEnv\lib\R\library\Rcpp\libs\x64\Rcpp.dll: Permission denied"
Warning message:
"restored 'Rcpp'"
package 'later' successfully unpacked and MD5 sums checked
Warning message:
"cannot remove prior installation of package 'later'"
Warning message in file.copy(savedcopy, lib, recursive = TRUE):
"problem copying C:\Users\ZLI2\Miniconda3\envs\myEnv\lib\R\library\00LOCK\later\libs\x64\later.dll to C:\Users\ZLI2\Miniconda3\envs\myEnv\lib\R\library\later\libs\x64\later.dll: Permission denied"
Warning message:
"restored 'later'"
The downloaded binary packages are in
C:\Users\ZLI2\AppData\Local\Temp\3\Rtmp6hrUI3\downloaded_packages
Running `R CMD build`...
* checking for file 'C:\Users\ZLI2\AppData\Local\Temp\3\Rtmp6hrUI3\remotes944065c13363\romanhaa-cerebroApp-0de48b6/DESCRIPTION' ... OK
* preparing 'cerebroApp':
* checking DESCRIPTION meta-information ... OK
* checking for LF line-endings in source and make files and shell scripts
* checking for empty or unneeded directories
Omitted 'LazyData' from DESCRIPTION
* building 'cerebroApp_1.3.1.tar.gz'
Old packages: 'ica', 'later', 'Rcpp'
cerebroApp::exportFromSeurat(nGene = "nFeature_RNA",
nUMI = "nCount_RNA",
groups = "seurat_clusters",
object = df0,
assay = "RNA",
file = "df0.crb",
experiment_name = "workshop",
organism = "Human")
[15:52:47] Start collecting data...
[15:52:47] Overview of Cerebro object:
class: Cerebro_v1.3
cerebroApp version: 1.3.1
experiment name: workshop
organism: Human
date of analysis:
date of export: 2022-07-10
number of cells: 2,125
number of genes: 16,514
grouping variables (1): seurat_clusters
cell cycle variables (0):
projections (1): umap
trees (0):
most expressed genes:
marker genes:
enriched pathways:
trajectories:
extra material:
[15:52:47] Saving Cerebro object to: df0.crb
[15:52:52] Done!
cerebroApp::launchCerebro()
##---------------------------------------------------------------------------##
## Launching Cerebro v1.3
##---------------------------------------------------------------------------##
Loading required package: shiny
Listening on http://127.0.0.1:6565
class: Cerebro_v1.3
cerebroApp version: 1.3.0
experiment name: pbmc_10k_v3
organism: hg
date of analysis: 2020-09-21
date of export: 2020-09-21
number of cells: 501
number of genes: 1,000
grouping variables (3): sample, seurat_clusters, cell_type_singler_blueprintencode_main
cell cycle variables (1): Phase
projections (4): tSNE, tSNE_3D, UMAP, UMAP_3D
trees (3): sample, seurat_clusters, cell_type_singler_blueprintencode_main
most expressed genes: sample, seurat_clusters, cell_type_singler_blueprintencode_main
marker genes:
- cerebro_seurat (3): sample, seurat_clusters, cell_type_singler_blueprintencode_main
enriched pathways:
- cerebro_seurat_enrichr (3): sample, seurat_clusters, cell_type_singler_blueprintencode_main,
- cerebro_GSVA (3): sample, seurat_clusters, cell_type_singler_blueprintencode_main
trajectories:
- monocle2 (2): all_cells, subset_of_cells
extra material:
- tables (1): SingleR_results
class: Cerebro_v1.3
cerebroApp version: 1.3.1
experiment name: workshop
organism: Human
date of analysis:
date of export: 2022-07-10
number of cells: 2,125
number of genes: 16,514
grouping variables (1): seurat_clusters
cell cycle variables (0):
projections (1): umap
trees (0):
most expressed genes:
marker genes:
enriched pathways:
trajectories:
extra material:
Warning message:
"`autoHideNavigation` only works with DT client mode and it will be ignored"
Warning message:
"The select input "expression_genes_input" contains a large number of options; consider using server-side selectize for massively improved performance. See the Details section of the ?selectizeInput help topic."
Warning message:
"`autoHideNavigation` only works with DT client mode and it will be ignored"
Warning message:
"`autoHideNavigation` only works with DT client mode and it will be ignored"
Warning message:
"`autoHideNavigation` only works with DT client mode and it will be ignored"
Warning message:
"`autoHideNavigation` only works with DT client mode and it will be ignored"